In the realm of medicine, there are countless fascinating conditions that captivate the attention of researchers and doctors alike.
One such enigmatic disorder is congenital adrenal cortical hyperplasia, a genetic anomaly with far-reaching effects on the adrenal glands.
Join us as we explore the intricate details of this condition, delving into its diagnostic methods, management strategies, and the incredible advancements in medicine that have transformed the lives of those affected by it.
Get ready to embark on a journey of discovery into the world of congenital adrenal cortical hyperplasia.
congenital adrenal cortical hyperplasia
Congenital adrenal cortical hyperplasia (CAH) is a group of genetic disorders that affect the adrenal glands.
CAH results in a lack of enzymes needed to produce hormones like cortisol, aldosterone, sex hormones, and adrenaline.
Classic CAH is typically diagnosed at birth and can present with ambiguous genitalia, low cortisol and aldosterone levels, and high levels of male hormones.
Nonclassic CAH is milder and often diagnosed later in life, with symptoms like excess body hair and irregular periods in women.
The most common cause of CAH is a genetic mutation in the 21-hydroxylase enzyme, resulting in increased production of male hormones.
Diagnosis can be done through prenatal testing or blood tests and genetic testing after birth.
Treatment aims to reduce excessive androgens and replace deficient hormones using medication like corticosteroids, fludrocortisone, and anti-androgen drugs.
Regular monitoring is necessary, and individuals with CAH may require information on managing their illness and potential surgical interventions for functional or cosmetic reasons.
Key Points:
- Congenital adrenal cortical hyperplasia (CAH) is a group of genetic disorders affecting the adrenal glands.
- CAH results in a lack of enzymes necessary to produce hormones like cortisol, aldosterone, sex hormones, and adrenaline.
- Classic CAH is typically diagnosed at birth and can present with ambiguous genitalia, low cortisol and aldosterone levels, and high levels of male hormones.
- Nonclassic CAH is milder and often diagnosed later in life, with symptoms including excess body hair and irregular periods in women.
- The most common cause of CAH is a genetic mutation in the 21-hydroxylase enzyme, leading to increased production of male hormones.
- Diagnosis can be done through prenatal testing or blood tests and genetic testing after birth, and treatment involves medication to reduce excessive androgens and replace deficient hormones.
congenital adrenal cortical hyperplasia – Watch Video
💡
Pro Tips:
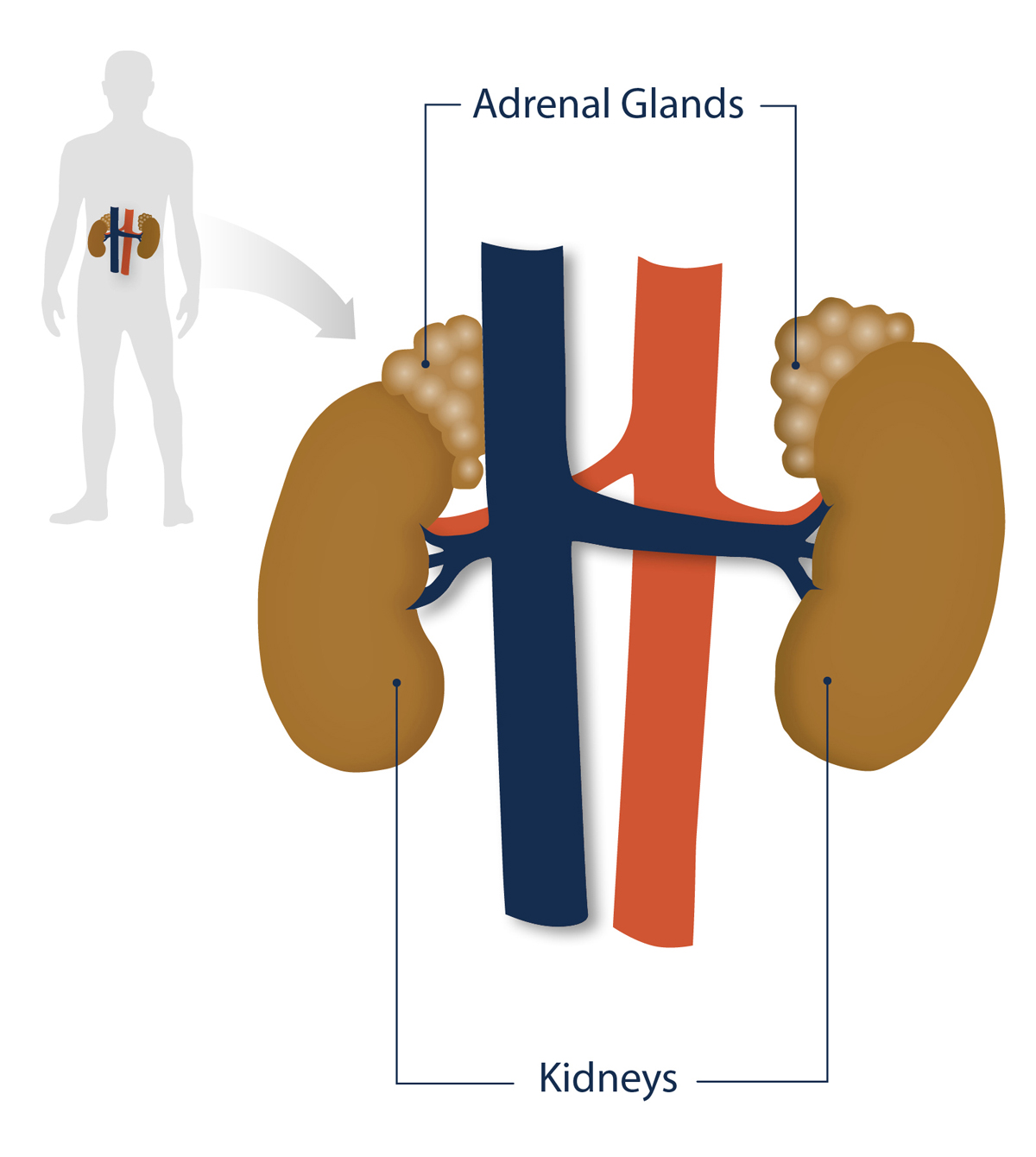
1. Congenital Adrenal Cortical Hyperplasia (CAH) is a rare genetic disorder that affects the adrenal glands, which are located just above the kidneys.
2. CAH is caused by a mutation in the CYP21A2 gene, which is responsible for producing enzymes that are crucial for the production of cortisol and aldosterone hormones.
3. CAH can lead to an imbalance of hormones, resulting in the overproduction of androgens (male sex hormones) in both males and females. This can lead to ambiguous genitalia in newborn females and early development of pubic hair in both sexes.
4. In some cases of CAH, individuals may experience salt wasting, which can lead to dangerously low levels of sodium in their bodies. This requires careful management of electrolyte levels to avoid severe complications.
5. Although CAH is a lifelong condition, with proper medical management and treatment, individuals can lead healthy and fulfilling lives. However, it may require lifelong hormone replacement therapy and regular monitoring to ensure optimal health.
Introduction To Congenital Adrenal Hyperplasia (CAH)
Congenital adrenal hyperplasia (CAH) is a group of genetic disorders that affect the adrenal glands, which are small, triangular-shaped organs located on top of the kidneys. These glands play a crucial role in producing hormones that are essential for normal bodily functions. However, individuals with CAH lack certain enzymes necessary for the production of these hormones, leading to imbalances that can have significant effects on their health.
In individuals with CAH, the enzymes responsible for producing hormones such as cortisol, aldosterone, sex hormones, and adrenaline are either deficient or non-functioning. As a result, there is a disruption in the normal hormonal balance, leading to a variety of symptoms and complications. The severity of CAH can vary, with some individuals experiencing milder forms of the condition, while others may have more severe symptoms from birth.
Enzyme Deficiency And Hormone Production In CAH
CAH is primarily caused by a deficiency in certain enzymes involved in the production of hormones. The most common form of CAH is caused by a genetic mutation in the 21-hydroxylase enzyme. This enzyme is responsible for converting cholesterol into cortisol and aldosterone, which are important hormones in the body. When the 21-hydroxylase enzyme is deficient, the body is unable to produce adequate amounts of these hormones.
Additionally, the lack of cortisol and aldosterone triggers a compensatory response in the body, leading to an increase in the production of androgens, which are male sex hormones. Consequently, individuals with CAH often have elevated levels of male hormones, which can result in a variety of symptoms.
Classic CAH: Diagnosis And Symptoms In Newborns
Classic Congenital Adrenal Hyperplasia (CAH) is typically diagnosed at birth or shortly thereafter. Newborns with classic CAH may present with ambiguous external genitalia, which can make it difficult to determine the individual’s sex at first glance. This is because the excess androgens produced as a result of the enzyme deficiency can cause virilization, or the development of male characteristics, in both males and females.
In addition to ambiguous genitalia, infants with classic CAH may also exhibit low cortisol and aldosterone levels, leading to symptoms such as:
- Poor feeding
- Dehydration
- Electrolyte imbalances
Additionally, the high levels of androgens can cause:
- Increased pigmentation of the genitalia
- Accelerated growth
- Premature development of pubic hair in both males and females.
Although classic CAH can present challenges in determining the individual’s sex at birth, the symptoms of low cortisol and aldosterone levels, along with the development of male characteristics, can provide important clues for diagnosis.
Nonclassic CAH: Diagnosis And Symptoms In Older Children And Adults
Nonclassic CAH is a milder form of the disorder that is often diagnosed later in childhood or early adulthood. Unlike classic CAH, individuals with nonclassic CAH may not exhibit ambiguous genitalia at birth. Instead, they may have more subtle symptoms that become apparent as they grow older.
Common symptoms of nonclassic CAH include excessive body hair and irregular periods in women. These symptoms are a result of the elevated androgens in the body, which can lead to hirsutism (increased hair growth) and menstrual irregularities. In some cases, nonclassic CAH may go undiagnosed until adulthood, as the symptoms may not be severe enough to warrant medical attention earlier in life.
Genetic Mutation And Its Role In CAH
The underlying cause of CAH is a genetic mutation that impacts specific enzymes involved in hormone production. In classic CAH, the most common form of the disorder, the mutation occurs in the gene responsible for encoding the 21-hydroxylase enzyme. This mutation results in a deficiency or complete absence of the enzyme, affecting the production of cortisol and aldosterone.
The genetic mutation associated with CAH is generally inherited in an autosomal recessive manner, which means both parents must carry and pass on the mutated gene for their child to develop the disorder. However, in some cases, the mutation may occur spontaneously, even without an affected family member.
Varied Symptoms Of CAH And Their Impact
The symptoms of CAH can vary depending on the specific gene defect and the degree of enzyme deficiency. In addition to the virilization of external genitalia in newborns and excessive body hair and irregular periods in older children and adults, other symptoms of CAH may include:
- Low appetite
- Nausea
- Dizziness
- Fatigue
One of the most serious complications of CAH is adrenal crisis. It is a potentially life-threatening condition characterized by a sudden and severe deficiency of cortisol and aldosterone. Some of the symptoms of adrenal crisis include:
- Low blood pressure
- Dehydration
- Electrolyte imbalances
- Loss of consciousness
Adrenal crisis requires immediate medical attention and treatment with intravenous fluids and corticosteroids.
Note: Adrenal crisis is a serious condition that should not be taken lightly. Immediate medical intervention is crucial to prevent further complications.
CAH In Males And Females
CAH affects both males and females, although the specific symptoms and their severity may differ between the sexes.
Males with CAH may experience abnormal genital development, such as an enlarged clitoris and fusion of the labia. In some cases, surgery may be considered to alter the appearance of the genitalia for both functional and cosmetic reasons.
Females with CAH usually experience disruption of normal menstrual cycles and the development of excessive body hair. Concerns for women with CAH may include irregular periods and fertility issues. Management of these symptoms often involves hormone replacement therapy and other medical interventions.
Diagnosing CAH Through Prenatal And Postnatal Testing
CAH can be diagnosed either prenatally or after birth, depending on various factors. Prenatal testing involves analyzing a sample of fetal cells or amniotic fluid to identify genetic mutations associated with CAH. This testing is usually recommended when there is a family history of the disorder or if certain risk factors are present.
After birth, CAH can be diagnosed through blood tests that measure hormone levels and genetic testing to confirm the specific gene defect. Newborn screening programs in many countries now include CAH testing, which allows for early detection and prompt treatment.
Treatment Options For CAH
The treatment of CAH aims to reduce excessive androgens and replace deficient hormones. This typically involves a combination of medication and ongoing monitoring to ensure optimal hormone levels and minimize the risk of complications.
The primary medication used in the treatment of CAH is corticosteroids, such as hydrocortisone or prednisone. These medications help to replace the missing cortisol and suppress the overproduction of androgens. Additionally, fludrocortisone, a synthetic form of aldosterone, may be prescribed to replace the deficient levels of this hormone.
In certain cases, individuals with CAH may also require salt supplements to maintain proper electrolyte balance and manage fluid imbalances. Oral contraceptive pills are sometimes prescribed to regulate menstrual cycles in women with CAH, while anti-androgen drugs may be used to reduce excessive hair growth.
Monitoring Hormone Levels And Managing CAH
Regular physical exams and blood tests are necessary for individuals with CAH to monitor hormone levels and adjust medication dosages accordingly. These tests help healthcare professionals to assess the effectiveness of treatment and detect any potential complications or hormone imbalances.
In addition to strictly adhering to medication regimens, individuals with CAH should receive education on managing their illness and recognizing signs of adrenal crisis. They should also be provided with information about surgical options, particularly for girls and women with CAH who may consider alterations to their genitalia for functional or cosmetic reasons.
Congenital adrenal hyperplasia is a group of genetic disorders that affect the adrenal glands and disrupt hormone production. The severity and symptoms of CAH can vary depending on the specific enzyme deficiency and gene defect involved. Fortunately, with early diagnosis and appropriate treatment, individuals with CAH can lead healthy and fulfilling lives with minimal complications. Regular monitoring and management of hormone levels are essential for maintaining optimal health and preventing adrenal crises.
💡
You may need to know these questions about congenital adrenal cortical hyperplasia
What is the life expectancy of someone with congenital adrenal hyperplasia?
The life expectancy of individuals with congenital adrenal hyperplasia (CAH) is not significantly affected by the condition. CAH is a well-understood diagnosis with effective treatment options readily accessible. People living with CAH lead healthy lives and have the freedom to pursue their aspirations, such as developing successful careers, building relationships, and starting families. With appropriate management and care, individuals with CAH can enjoy a normal life expectancy without any notable impact on their overall well-being.
How serious is congenital adrenal hyperplasia?
Congenital adrenal hyperplasia (CAH) is a serious condition that can have significant implications on both males and females. One form of CAH, Steroid 3-Beta-hydroxysteroid dehydrogenase deficiency, can result in ambiguous genitalia in both genders, leading to challenges in gender identification. Additionally, this form of CAH may also lead to salt-wasting, further complicating the condition.
Another form of CAH, called congenital lipoid adrenal hyperplasia, presents a particularly serious threat. This condition may cause early death due to adrenal crisis, highlighting the severity of the condition. In males, ambiguous genitalia is also a characteristic feature of this form of CAH. Overall, congenital adrenal hyperplasia presents significant health risks and requires prompt medical attention to ensure appropriate management and minimize complications.
What is the most common cause of congenital adrenal hyperplasia?
The most common cause of congenital adrenal hyperplasia (CAH) is a deficiency in the enzyme 21-hydroxylase. This enzyme is crucial for the production of important hormones in the body. Although there are other rare enzyme deficiencies that can also lead to CAH, 21-hydroxylase deficiency is the most prevalent and frequently associated with the condition.
What does congenital adrenal hyperplasia do?
Congenital adrenal hyperplasia (CAH) is a genetic disorder that affects the adrenal glands, leading to overproduction of androgens in the body. This excessive production of androgens can result in various complications. For newborn girls, it may lead to atypical genitalia, causing physical differences in the appearance of the external genitalia. In older children, CAH can cause early puberty, resulting in rapid growth and potentially contributing to shorter adult height. Additionally, individuals with CAH may face difficulties with fertility as a result of hormonal imbalances caused by the condition.
Reference source
https://www.hopkinsmedicine.org/health/conditions-and-diseases/congenital-adrenal-hyperplasia
https://www.rch.org.au/endo/cah/The_nature_of_CAH/
https://rarediseases.org/rare-diseases/congenital-adrenal-hyperplasia/
https://www.mayoclinic.org/diseases-conditions/congenital-adrenal-hyperplasia/symptoms-causes/syc-20355205